* E két szerző egyenlő mértékben vett részt a munkában
Egy in vitro iszkémia-reperfúzió modell eredményei
A koronáriabetegség eredményesebb kezelésének köszönhetően napjainkban, számos országban a szívelégtelenség gyakoribbá válása figyelhető meg. Jövőbeni lehetséges kezelés az orvostudomány jelenleg legígéretesebb és leginkább kutatott területe, az őssejtes terápia. A beadott sejtek jótékony túlélése, és ennek kapcsán hatása azonban nem elégséges, feltehetően az érintett területen uralkodó oxidatív környezet miatt, mely fokozott pusztulásukhoz vezet. Ilyen környezetben már számos más esetben igazolást nyert a poli(ADP-ribóz) polimeráz (PARP) nukleáris enzim gátlásának előnyös hatása. Ezért feltételeztük, hogy a terápiásan hozzáadott sejtek PARP inhibitoros kezelése eredményesebb túlélésükhöz, és így hatásuk felerősítéséhez vezet egy in vitro szimulált iszkémiareperfúzió modellben. Az iszkémiás körülményeket oxigén- és glükóz megvonásával hoztuk létre, majd különböző koncentrációjú PARP inhibitorral előkezelt sejteket adtunk a posztiszkémiás sejtekhez. A terápiásan hozzáadott sejtek túlélése szignifikánsan emelkedett a PARP gátlás hatására, továbbá az iszkémia-reperfúziót elszenvedett sejtek túlélési aránya is javulást mutatott a PARP inhibitorral előkezelt őssejtek hatásának köszönhetően. Ennek alapján a terápiásan hozzáadott sejtek prekondicionálása PARP inhibitorral jótékonyan befolyásolja a sejtalapú terápiák hatékonyságát. Ez a módszer, a sejtalapú terápiák lehetőségének hatékonyabb kihasználását jelentheti a jövőben.
Érbetegségek: 2013/2. 41-50. oldal
KULCSSZAVAK:
szívinfarktus, szívelégtelenség, őssejtes terápia, poli(ADP-ribóz) polimeráz
Egy in vitro iszkémia-reperfúzió modell eredményei
Bevezetés
Európában jelenleg a szív- és érrendszeri megbetegedések a halálesetek közel feléért felelősek. Ennek alapján megközelítőleg minden második ember halálát koszorúér megbetegedés, és minden harmadikét stroke okozza [1]. A koronáriabetegségben szenvedők száma az utóbbi 30 évben Észak- és Nyugat-Európában csökkenni kezdett, de Közép- és Kelet-Európában továbbra is erősen növekszik. A WHO legfrissebb hozzáférhető adatai szerint a 75 éves koruk előtt elhunyt férfiak körében 38%, míg a nőknél 42% volt a szív- és érrendszeri betegség következtében elhalálozottak aránya [1]. A miokardiális infarktus egyre eredményesebb kezelése ellenére sem valósul meg a károsodott szívizom funkciójának vissza - állítása, ezért a betegek jelentős részében hosszú távon szívelégtelenség alakul ki. A funkcionális regenerációt létrehozó sejtalapú terápiák alkalmazása - kombinálva a konvencionális módszerekkel - jelentheti a jövőt ezen betegek kezelésében.
Sejtalapú terápiák
Napjainkban az őssejtek terápiás alkalmazása a biológia és az orvostudomány egyik leginkább kutatott és legígéretesebb területe. Az egyik lehetséges felhasználási módot a szívinfarktust követő őssejtbeültetés jelenti [2]. A sejttranszplantációval történő szívizom-regeneráció új távlatokat nyitott az infarktus kezelésében, és több különböző kutatás is bebizonyította, hogy ennek a megközelítésnek valóban kedvező hatása van a sérült szív funkciójának fokozásában [3, 4]. Azonban közel ezer ischaemiás szívbetegségben szenvedő személy vizsgálata kimutatta, hogy a csontvelői eredetű sejtek transzplantációja mindössze 3,66%-os javulást mutatott a bal kamrai ejekciós frakcióban [5-7]. Ez az eredmény jóval az új terápiához fűzött elvárások alatt volt. Ez a kiábrándító adat a sejtalapú terápiák tisztázatlan hatásmechanizmusának lehet következménye. A mechanizmust tekintve a legvalószínűbb feltételezés az, hogy a sejtek parakrin módon befolyásolják a környezetüket, és így hoznak létre pro-angiogenetikus és anti-inflammatorikus hatásokat, de elképzelhető a sejtek direkt kölcsönhatása is [8-12]. Megjegyzendő továbbá, hogy a legtöbb terápiásan hozzáadott sejt elpusztul a posztiszkémiás miokardium agresszív környezetében [13-15]. Egyes irodalmak szerint a terápiásan alkalmazott sejtek hatása kimerül abban, hogy apoptózissal elpusztulva visszafogják a gyulladásos folyamatokat [16], és ez a jelenség akár terápiásan is alkalmazható lehet [17]. A beadott sejtek differenciálódása a célszerv sejttípusává az irodalom alapján nem történik meg, azonban ritkán sejtfúzió tapasztalható [18]. A jelenlegi terápiás megoldások ezért lényegében a mikrokörnyezet befolyásolását tűzik ki célul, elsősorban parakrin mediátorok által. A parakrin faktorok mellett a mikrokörnyezet befolyásolásában szerepe lehet a sejtek között kialakuló nanocsöveknek is, amelyeken keresztül mitokondriumok és egyéb sejtorganellumok áramlását figyelték meg [19]. A mechanizmusok aránya a regeneráció során is feltehetően dinamikusan változik. Kezdetben a sejtek parakrin gyulladáscsökkentő és anti-apoptotikus hatása, később az érképzés és az elpusztult szívizomszövet pótlása és regenerálása lehet a legfontosabb. A mechanizmusokat tekintve feltételezhető, hogy a hozzáadott sejtek túlélésének növelése a terápia hatékonyságának növekedésével járna együtt, akár parakrin úton, akár sejt-sejt közötti kapcsolatok által fejtik ki. E feltevés a különböző előkezelések hatására elért jobb túléléssel már igazolást nyert [20-23].

1. ábra.
A PARP működése.
Poli (ADP-riboz) polimeraz (PARP)
Az infarktus kezelese soran kialakulo reperfuzio nemcsak oxigent es glukozt, hanem feherversejteket is szallit az erintett teruletre, s utobbiak szamos gyulladaskelt. faktort, tobbek kozott interleukinokat es szabad gyokoket bocsatanak ki a serult szovettel talalkozva. A makrofagok interleukinok hatasara nitrogen-oxid szintazt bocsatanak ki, ami L-arginin szubsztratbol nitrogen-monoxidot (NO) termel. A keletkezett nitrogen-monoxidbol a mikro - kornyezett.l fugg.en kulonboz. reaktiv nitrogen vegyuletek keletkezhetnek: nitrozium kation (NO+), nitroxil anion (NOP), illetve szuperoxiddal reagalva a kulonosen reaktiv peroxinitrit (ONOOP). A szabad gyokok a sejteket karositjak azaltal, hogy DNS-torest, koros feherjeszerkezetet, lipidperoxidaciot okoznak. Az oxidativ DNS serules poli(ADP-riboz) polimeraz-1 (PARP) nukleáris enzim aktivációját hozza létre. A PARP egy nukleáris enzim, mely DNS-törések felismerésére és javítására képes. Kis noxa esetén a PARP aktivációja segíti a sejt túlélését, a DNS-repair mechanizmusát, nagy noxa esetén azonban a NAD+ elhasználásával és az ATPszintézis csökkentésével sejtnekrózishoz vezet (1. ábra).
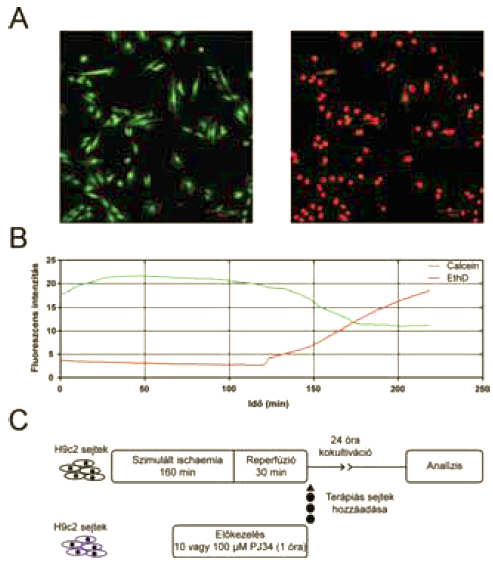
2. ábra.
A kísérleti metodika. A) Konfokális mikro szkópos kép az oxigén- és glükózmegvonás előtti és utáni állapot összehasonlítására. (Calcein-AM – zöld; ethidium-homodimer - vörös).
B) Élő-halott festéssel összefüggő fluoreszcencia-intenzitások változása szimulált ischaemia alatt.
C) Kísérleti protokoll.
A PARP gyakorlati jelentősége
Az ADP-ribóz polimerek akkumulációja figyelhető meg stroke-ban, infarktust követően, szív transzplantációjakor, valamint a bélrendszerben, a tüdőben és a szívben szepszis vagy hemorrhagiás sokk esetén. Emelkedett PARP aktivitás figyelhető meg a reperfúziót követő 2. és 24. óra között, ez a reaktív oxigén és nitrogénvegyületek hosszan tartó jelenlétére utal a szívizomban [24]. Jellemzően a nekrotikus zónában és a "veszélyeztetett területen" a legmagasabb az aktiváció. Számos in vivo és in vitro modellben kimutatták, hogy az enzim gátlásával tulajdonképpen direkt módon gátolható a celluláris energiaháztartás összeomlása, ezáltal a szívizomsejtek nekrózisa a peri-infarktusos zónában. Knock-out egerekben a PARP funkcionális génjének hiánya protektívnek bizonyult a reperfúziós károsodással szemben.
A különböző PARP inhibitorokkal végzett kísérletek során az infarktus által érintett zóna mérete csökkent, megőrződött az ATP-készlet és csökkent a kreatinfoszfokináz szintje [25, 26]. Stroke-ban játszott szerepét állatkísérletekkel támasztották alá, PARP knock-out egerekben a középagyi artéria tranziens okklúziója után jelentősen csökkent infarktusméret volt megfigyelhető, és ez kizárólag a PARP-1 gén termékének hiányával volt magyarázható [27]. Haddad és mtsai Swiss hím egerekben a bal arteria cerebri media 1 órás okklúziójával agyi infarktust okoztak. A PARP-inhibitor PJ34 csökkentette a TNFα plazmaszintjét 6 óra elteltével, továbbá az mRNS-ek vizsgálatakor mind a TNFα, mind az IL-6, E-szelektin és az ICAM-1 molekulák esetében is szignifikáns csökkenést találtak [28]. Egy 2004-es tanulmányban azt találták, hogy a 90 perces a. cerebri media okklúzió után a PARP szint emelkedett és a NAD+ mennyiségének csökkent 1 órás reperfúziót követően. Ugyanakkor PARP-inhibitor csökkentette e káros értékeket 24 órával az infarktust követően. [29].
A PARP aktiváció kapcsolatba hozható a szisztémás gyulladással és a keringési sokkal is, mely összefügg az emelkedett oxigéngyök szinttel továbbá az iNOS túlexpresszióval. Az NO és szuperoxid egymással reagálva peroxinitritet hoz létre, és mind a három forma szerepet játszik a kardiális diszfunkció és sokszervi elégtelenség patogenezisében. A peroxinitrit a sokkhoz hasonló patofiziológiai változásokat képes létrehozni, és ezek a változások részben összefüggést mutatnak a PARP aktivációval. A PARP gátlás ilyen körülmények között is előnyösnek mutatkozott [27]. Virág és mtsai immunhisztokémiai úton detektálták, hogy a PARP aktivitás már 2 órával a reperfúzió megkezdődése után nőtt a reperfundált miokardiumban, s további 24 óráig emelkedett marad. A legmagasabb PARP aktiváció a nekrózis területén, illetve a peri-infarktusos zónában volt mérhető. Festési eljárásokkal azt találták, hogy a legnagyobb PARP aktivitás a szívizomsejtekben van, és nem az odavándorló mononukleáris sejtekben. Megállapításuk szerint mivel a PARP aktiváció indukálja a sejtnekrózist a celluláris energetika összeomlása által, a PARP inhibitorok kardioprotektív szerepét ki lehetne használni a terápiában [27]. Egy további kutatási irányt jelent az iszkémiás prekondicionálás, mely közismerten védelmező hatással rendelkezik a szívizom iszkémiás-reperfúziós sérüléseiben. Ennek mechanizmusa minden bizonnyal összefüggésben van a PARP aktivációjával. A prekondicionálás alatt a sejtek kisebb koncentrációban találkoznak reaktív oxigén- és nitrogén vegyületekkel, amelyek mérsékelt PARP aktivációt váltanak ki, vélhetően a PARP autoribozilációját is kiváltva, mely az enzim gátlásához vezet. A későbbi reperfúziós sérülés alatt a nem kezelt egyedekhez képest csillapított PARP aktiválódás következik be, csökkentve a későbbi káros következményeket. Fiorillo és mtsai PARP inhibitorral (PJ34) végzett kísérleteikben a gátlószert az iszkémia-reperfúziós károsodást szenvedett sejtekhez adták a reoxigenizáció kezdetekor, s vizsgálták az oxidativ stressz, a PARP-1, NAD+ és ATP depléciót, LDH szintet. A mért paraméterek mindegyike csökkent a PJ34 szerrel kezelt csoportban [30].
Ezekben a vizsgálatokban a postiszkémiás sejtekhez adták a gátlószert, viszont a sejtterápia esetén a nagy kihívást az jelenti, hogy a sejttranszplantációt követően a sejtek nagy része elvész a rossz véráramlás, az iszkémia-reperfúziós károsodás és a gyulladásos faktorok miatt. Ezért kísérleteink célja in vitro körülmények között szimulált iszkémia-reperfúzió (I-R) modellben annak vizsgálata, hogy növeli-e a hozzáadott sejtek PARP gátlása a hozzáadott sejtek életképességét, illetve a posztiszkémiás sejtek életképességét.
Anyagok és módszerek
Felhasznált sejttípusok
Kísérleteinkhez patkányból származó H9c2 kardio - mioblaszt sejtvonalat (ATCC, Wesel, Németország) használtunk. A kardiomioblasztokat 4,5 g/l glükóztartalmú DMEM-ben tenyésztettük, amely tartalmazott még 10% FCS-t, 2 mM L-glutamint, 100 U/ml penicillint és 100 µg/ml streptomycint, 37°C-on és 5%-os CO2 környezetben. A sejttenyészetre tett médiumot 2-3 naponta cseréltük, és a Petri csészék 70-80%-os konfluencia - szintjénél passzáltuk a sejteket. A kísérletekhez 7 és 13 közötti passzázsszámú sejteket használtunk. A H9c2 kardiomioblaszt sejtvonal embrionális patkány szívből származik, szívizomra és vázizomra is jellemző elektrofiziológiai és biokémiai tulajdonságokat mutat, ezért mind szívizom, mind vázizom in vitro modelljeként használatos [31].
In vitro iszkémia-reperfúzió modell
In vitro patkány H9c2 kardiomioblaszt kultúrában az oxigén és glükóz megvonása iszkémiás jellegű viszonyok kialakulásához vezet. A sejtek inkubációja glükózmentes médiumban, 0,4% oxigén (3,25 Hgmm) és 99,6% nitrogén összetételű atmoszférában zajlott 160 percen keresztül (PECON inkubációs rendszer). Az inkubációs rendszer a konfokális mikroszkóp része, segítségével szabályozható a hőmérséklet és az O2 koncentráció. A szimulált iszkémia optimális időtartamát kísérleti úton határoztuk meg. A két napos protokoll első napján a H9c2 sejteket 42 mm-es tárgylemezre tettük ki, majd egy napig 37°C-os, 5%-os széndioxid szintet biztosító inkubátorban növesztettük. Az egy napos várakozás oka az volt, hogy a sejtek biztosan letapadjanak az új felszínhez, s az új környezetet megszokva, ,,normál” funkciót (növekedést, sejtciklust, sejt-sejt kapcsolatot) mutassanak. A második napon történt az oxigén és glükóz megvonás, melynek során a glükózmentes médium calcein és ethidium-homodimer festékeket tartalmazott. Konfokális mikroszkóppal 1 kép/perc gyakorisággal felvételeket készítettünk és a két fluoreszcens festék intenzitás változását követtük nyomon. Végpontnak azt az állapotot vettük, amikor az összes sejt kizárólag EthD festődést mutatott (2A és 2B ábrák). A fluoreszcenciák időbeli lefutását értékelve kiválasztottuk azt az időpontot (160 perc), amikor a sejtek megközelítőleg fele, nekrotizált [12].
In vitro sejtalapú terápia modell
A terápiás céllal alkalmazott H9c2 sejteket 3 kísérleti csoportba osztottuk és 1 órás előkezelésnek vetettük alá: (1) fiziológiás sóoldattal kezelt kontroll csoport (2) 10 µM PJ34-el kezelt csoport (PARP inhibitor, Inotek Pharmaceuticals Corp., Beverly, MA, USA), (3) 100µ PJ34-el kezelt csoport. Az előkezelést követően a H9c2 sejteket kétszer mostuk PBS-sel, hogy a PARP-gátlóból ne maradjon az oldatokban, majd a sejteket feltripszineztük a Petri-csészékből, centrifugálással összegyűjtöttük, és 20 000 sejtet adtunk a 12 lyukú lemez minden egyes kompartmentjébe, amelyekben 30 000 "posztiszkémiás" sejt esett át a szimulált ischaemia-reperfúzión. A sejteket további 24 órán át inkubátorban tartottuk, majd laktátdehidrogenáz- felszabadulási és metabolikus aktivitási méréseket, továbbá áramlási citometriás méréseket végeztünk (2C. ábra).

3. ábra.
Reprezentatív kép a 3 sejtcsoportról a sejttúlélés vizsgálata során (zöld: calcein-AM pozitív élő sejtek; piros: ethidium-homodimer-2 pozitív nekrotikus sejtek; lila: kettősen pozitív apoptotikus sejtek).
Laktát-dehidrogenáz és malondialdehid szint mérése
A felhasznált iszkémia-reperfúzió modellt ellenőriztük és a sejtek környezetét, illetve károsodását különböző vizsgálatokkal mértük fel. Az LDH mérés elve, hogy a sejtek nekrózisának mértéke, vagy egy adott anyag citotoxicitása meghatározható a membrán integritását vizsgáló módszerek segítségével. A laktát-dehidrogenáz (LDH) alapvetően intracelluláris enzim, extracellulárisan abban az esetben fordul elő, ha a sejtmembrán sérülésein keresztül képes elhagyni a sejtet. Számos más cito - plazmatikus enzimmel ellentétben stabil, nagy mennyiségben van jelen, és a plazmamembrán sérülését követően azonnal detektálható a sejtkultúra felülúszójában.
Az LDH aktivitás egy párosított enzimreakció segítségével határozható meg, melynek során az LDH a laktátot piruváttá oxidálja és közben redukált koenzim (NADH) képződik. A redukált koenzimet a NADH dehidrogenáz oxidálja, eközben az indonitrotetrazóliumból formazan képződik. A formazán mennyiségének növekedése közvetlenül korrelál a lizált sejtek számával, és spektrofotométerrel mérhető.
Az LDH kibocsátást három különböző ok miatt vizsgáltuk, egyrészt az iszkémia-reperfuziós modellünk értékelésére, másrészt, hogy meghatározzuk a PJ34 citotoxicitását és hatásosságát a H9c2 sejteken, harmadrészt a kísérletes csoportokban a nekrózisos sejtek számának összehasonlítására. A modell értékelésére 100 000 H9c2 sejtet használtunk és 24 órával a reperfúzió kezdete után végeztünk méréseket. A PJ34 PARP inhibitor hatásosságának vizsgálatára mértünk két sejtcsoportot melyek 10 µM illetve 100µM PJ34-el voltak előkezelve és egy kontroll csoportot, melyen nem történt előkezelés (2C. ábra). Inkubáció után a különböző csoportokból 10 000 sejtet helyeztünk a 12 lyukú lemez minden egyes kompartmentjébe és mindegyik csoportot vagy vehikulummal kezeltük, vagy 400 µM H2O2-t raktunk a sejtekre 2 órára, hogy az inhibitor hatékonyságát vizsgáljuk. A sejtek ezt követően lizálásra kerültek 1% Triton-X-el vagy sejtlizáló oldattal, hogy megkapjuk a háttér és a teljes LDH tartalmat. A harmadik esetben 24 órával a reperfúzió után a sejtek felülúszójában mértük a teljes LDH szintet és összehasonlítottuk a vehikulummal kezelt csoport (H9c2) értékeivel. Az abszorbanciát 450 nm és 650 nm-en mértük és ebből számoltuk a citotoxicitás mértékét. A nekrózis mértéke mellett az oxidatív stressz mértékét is meg kívántuk határozni, ezért malondialdehid mérést végeztünk a felülúszóból. Az utóbbi években számos in vivo és in vitro módszert dolgoztak ki a malondialdehid kimutatására [32].
A malondialdehid (MDA) szint a szimulált iszkémiareperfúziós modellben keletkezett lipidperoxidáció mértékét mutatja meg és tiobarbiturát savra reaktív anyaggal mérhetővé tehető. A mérési protokoll limitációja miatt itt 1 000 000 sejtet használtunk. 5 órával a szimulált reperfúzió kezdete után 50µl-t a sejtek felülúszójából egy reaktív elegyhez adtunk, mely 50µl 8,1% sodium dodecyl szulfátot, 375µl 20%-os ecetsavat (pH 3,5) és 150 µl desztillált vizet tartalmazott. A keverék 375 µl frissen előállított, forrási hőmérsékletű tiobarbiturát sav (0,8%) hozzáadásával lett teljes, majd 95°C -on egy órán keresztül inkubáltuk. Szobahőmérsékletre való lehűtés után, 200 µl felülúszót 96 lyukú microplatekbe osztottuk szét, majd 532 nm-en mértük az abszorbanciát.
Áramlási citométriás mérések
Áramlási citometriával 24 órával a reoxigenizáció után vizsgáltuk a sejteket (FACSCaliburTM, Becton Dickinson, Franklin Lakes, NJ, USA). Ennek során a sejteket tripszinálás után reszuszpendáltuk 250 nM calcein-AM (ex/em: 494/517 nm, Invitrogen, Carlsbad, CA, USA), és 350 nM ethidium-homodimer-2 (ex/em: 536/624 nm, Invitrogen, Carlsbad, CA, USA) tartalmú 500 µl PBS oldatban. Az áramlási citometria segítségével el tudtuk különíteni a terápiásan hozzáadott sejteket a postiszkémiás sejtektől Vybrant DiD jelölésük alapján (ex/em: 633/665 nm; Molecular Probes, USA), és a megfelelő sejtek további analízisre kerültek. A sejthalál mértékének felmérése élő-halott festés alapján történt [33]. Összefoglalva, a maximálisan ethidium-homodimer-2 pozitív, és minimálisan calcein-AM negatív sejteket tekintettük nekrotikusnak, az ethidium-homodimer-2 negatív de calcein-AM pozitív sejteket pedig élőnek, illetve egy külön csoportját a sejteknek melyek közepesen festődtek, apoptotikusnak (3. ábra) Az áramlási citometriás mérések kiértékelése a Weasel programmal történt (WEHI, Ausztrália)

4. ábra.
A kísérletes modell validálása. A) A lipid-peroxidáció termék MDA koncentrációja a felülúszóban (***: p<0,001 átlag±SEM, n=6);
B) A sejtnekrózis százalékos aránya az LDH mérés alapján (** p<p<0,01 átlag±SEM, n=12);
C) A PJ34 citotoxicitása és a PARP-gátlás hatékonysága (***:p<0,001 átlag±SEM, n=6).
Anyagcsere-intenzitás
A sejtproliferáció és anyagcsere-intenzitás kvantitatív mérése az élő sejtek redukáló képességét felhasználva lehetséges. Számos módszer ismert az irodalomban, amely ezen az elven működik, mi a PrestoBlue reagenst választottuk méréseinkhez. Az oldat membránpermeábilis, kék színű resazurin komponenst tartalmaz, amely nem fluoreszcens. Az élő sejtekben piros színű erősen fluoreszcens resorufin vegyületté redukálódik. Spektrofotometriás kiértékelés esetén az abszorbancia eltolódása mérhető 600 nm-ről (resazurin) 570 nm-re (resorufin).
Statisztika
Az adatok statisztikai elemzése során varianciaanalízist és t-próbát alkalmaztunk a csoportok számától függően. Az adatok átlag±standard hiba(SEM) formában kerültek megadásra. A p<0,05 érték esetén tekintettük a különbségeket statisztikailag szignifikáns eltérésnek.
Eredmények
Szimulált iszkémia-reperfúziós protokoll
Az MDA vizsgálaton alapuló oxidatív stressz szint szignifikánsan emelkedett az iszkémia-reperfúziós modellben (I-R: 13,70±0,81 µM) a nem kezelt kontroll sejtekhez képest (0,47±0,18 µM 4A ábra). Az oxigén és a glükóz megvonása az LDH-szint szignifikáns emelkedés - éhez vezetett a felülúszóban a kontroll körülményekhez képest (kontroll: 0,00±0,81%; I-R model: 29,58±6,21%; 4B. ábra). Az oxigén és a glükóz megvonása az LDH-szint szignifikáns emelkedés - éhez vezetett a felülúszóban a kontroll körülményekhez képest (kontroll: 0,00±0,81%; I-R model: 29,58±6,21%; 4B. ábra). A PJ34 PARP inhibitor a használt koncentrációkban nem fejtett ki citotoxikus hatást a H9c2 sejtekre és szignifikánsan megvédte ezeket a sejteket a H2O2 által indukált sérülésektől (kontroll + H2O2: 35,14±1,01%; 10 µM PJ34 + H2O2 : 15,65±0,95%; 100 µM PJ34 + H2O2: 15,69±0,54%; 4C. ábra).

5. ábra.
Áramlási citometriás élő-halott analízis eredményei
A) Az élő, apoptotikus és nekrotikus sejtek százalékos arányai a terápiásan hozzáadott sejtekben (*:p<0,05 vs H9c2, **:p<0,01 vs H9c2, átlag±SEM, n=18);
B) Az élő, apoptotikus és nekrotikus sejtek százalékos arányai a postischemiás sejtekben (*:p<0,05 vs H9c2 , **: p<**: 0,01 vs H9c2 , ###: p<0,001 vs I-R modell, átlag±SEM, n=18).
Áramlási citometriás mérések
A terápiásan hozzáadott sejtek áramlási citometriás vizsgálata az élő, az apoptotikus és a nekrotikus populációiban is szignifikánsan nagyobb túlélést mutatott a PARP inhibitoros előkezelés esetén (H9c2: 52,02±5,01%, H9c2 + 10 µM PJ34: 63,38±4,50%, H9c2 + 100 µM PJ34: 64,99±3,47%), továbbá a PJ34 előkezelésnél a nekrotikus sejtek aránya csökkent (H9c2: 37,23±4,40%, H9c2 + 10 µM PJ34: 26,83±3,49%, H9c2 + 100 µM PJ34: 24,96±2,43%). Az apoptotikus sejtek arányában nem volt eltérés (H9c2: 10,87±1,12%, H9c2 + 10 µM PJ34: 9,22±1,28%, H9c2 + 100 µM PJ34: 10,18±1,55%; 5A ábra). A beállításoknak megfelelően nem volt szignifikáms különbség az I-R modell és a H9c2 kezelt csoport között az élő sejtek (I-R modell: 36,44±5,05%, H9c2: 42,81±5,11%) és nekrotikus sejtek (I-R modell: 43,64±4,00%, H9c2: 37,29±4,55%) arányát tekintve. Fontos azonban, hogy a posztiszkémiás sejtek túlélése magasabb volt, amikor PARP gátlóval kezelt terápiás sejteket használtunk (H9c2 + 10µM PJ34: 52,07±5,80%, H9c2 + 100 µM PJ34: 54,95±5,55%) és a nekrotikus sejtek aránya szintén csökkent (H9c2 + 10 µM PJ34: 30,18±4,60, H9c2 + 100 µM PJ34: 25,52±3,47%). Az apoptotikus sejtek arányában nem volt statisztikai különbség a csoportok között (kontroll: 19,94±2,75%, H9c2: 20,23±2,62%, H9c2 + 10 µM PJ34: 17,20±2,42%; H9c2 + 100µM PJ34: 20,05±3,23%; 5B. ábra).
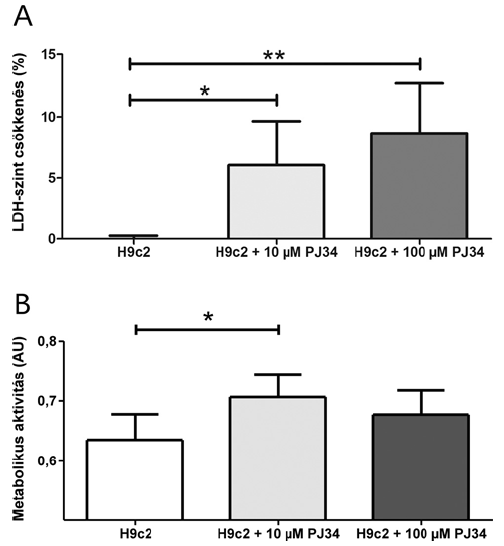
6. ábra.
Citotoxicitás és életképesség mérése.
A) Laktát-dehidrogenáz kibocsátás csökkenése invertált skálán (*:p<0.05, **:p B) Metabolikus aktivitás mérése PrestoBlue reagenssel (*:
Laktát dehidrogenáz (LDH) citotoxicitás vizsgálat
A terápiás sejtek PARP gátlóval történő előkezelése esetén az LDH szint szignifikánsan csökkent. 10 µM és 100 µM PARP inhibitort használva az LDH kibocsátás 6,04±3,61%, illetve 8,68±3,98%-kal mutatott alacsonyabb értéket (6A. ábra).
Metabolikus aktivitás
A 10 µM PJ34 használatával létrehozott PARP-gátlás szignifikánsan serkentette a sejtek összmetabolikus aktivitását (H9c2: 0,64±0,04; 10 µM PJ34: 0,71±0,04; önkényes egység). A100 µM PJ34-el előkezelt szívizomsejtek esetén csak egy nem szignifikáns trend volt kimutatható (0,68±0,04; 6B. ábra).
Diszkusszió
Tanulmányunk során kimutattuk, hogy a terápiás sejtek PJ34-gyel történő előkezelése javítja a túlélésüket, és növeli a posztiszkémiás sejtek életképességét is az általunk alkalmazott in vitro iszkémia-reperfúziós modellben.
A modellt először az oxidatív stressz, nekrotikus tulajdonságok vizsgálatára kalibráltuk, majd megnéztük a PARP-gátlószerünk citotoxicitását és hatásosságát. Azt találtuk, hogy az oxigén és a glükóz megvonásával párhuzamosan nőtt az MDA-szint (ahogy ez in vivo is megfigyelhető), illetve az LDH-méréssel kimutatható membránkárosodás alakult ki. Így az alkalmazott modellünk megfelelően szimulálja az iszkémia-reperfúziós sérülést. A sejtek hozzáadásának időpontját részben irodalmi adatok és részben saját kezdeti vizsgálataink alapján választottuk, utóbbiak azt sugallták, hogy eredményesebb a sejtterápia, ha a reperfúzió kezdetét követő 30 perc elteltével adjuk be a sejteket.
A PARP inhibitoros előkezelés hatásosságát áramlási citométerrel vizsgáltuk. A terápiás sejtek PARP inihbitorral való előkezelése javítja saját túlélésüket, ekkor nő a túlélő posztiszkémiás sejtek mennyisége is. Ugyanakkor kezeletlen terápiás sejtek adása nem volt szignifikáns hatással ezen a sejtpopuláción. Így arra következtethetünk, hogy az előkezelés miatti nagyobb túlélő terápiás sejtszám okozza a posztiszkémiás sejtek javuló életképességét. Ez a hatás azonban nem lehet a PARP-gátló kezelés hatása a posztiszkémiás sejtekre, hiszen az előkezelés a terápiás sejtek posztiszkémiás sejtekhez adása előtt történt, melyet egy médiumos átmosás követett, így a PARP inhibitor nem juthatott át a posztiszkémiás sejtekhez. A PJ34 védő hatása tehát a kezelés után is tart, és az elhúzódó PJ34 hatása nem kapcsolódik az inhibitor folyamatos jelenlétéhez, de kapcsolatban állhat a sérülés okozta pozitív feed-back mechanizmusok végleges megszakításával.
In vivo a hozzáadott sejtek az infarcerálódott terület nagyságát csökkenthetik. Korábbi vizsgálataink [12, 34] és más kutatócsoportok eredményei [10, 35] alapján úgy gondoljuk, hogy ez a kedvező hatás kapcsolatos lehet részben a sejt-sejt kapcsolatokkal és részben a terápiás sejtekből felszabaduló parakrin faktorokkal.
Korábbi tanulmányok azt mutatják, hogy a PARP inhibitorok gátolják az adhéziós receptorok expresszióját és a mononukleáris sejtek infiltrációjának mértékét csökkentik, így gátolva az intracelluláris prooxidánsok termelését [27]. Fontos megjegyezni, hogy mivel a PARP inhibitor blokkolja a DNS-javító mechanizmusokat, ezért felmerült, hogy potenciálisan genotoxikus lehet. Ezt azonban korábbi vizsgálatok cáfolják, egy tanulmány kimutatta, hogy a poli(ADP-ribozil)áció gátlása nem módosítja a genotoxicitást [36], míg egy másik bakteriális reverz mutációs teszttel, in vitro kromoszóma aberráció teszttel és csontvelő micronucleus teszttel arra a következtetésre jutott, hogy a PARP gátlás nem genotoxikus [37].
Kísérleteinkben a PARP gátlással történő előkezelés hatását LDH és PrestoBlue-festődés mérésekkel vizsgáltuk. Ez a két módszer tükrözi mindkét sejtpopuláció nekrózisának mértékét és metabolikus aktivitását a sejtek hozzáadását követően 24 óra elteltével. Az LDH értékek jelentősen csökkentek mindkét alkalmazott koncentrációjú PJ34 előkezelés esetében. Az előkezelés 10 µM PJ34-gyel szignifikánsan növelte a metabolikus aktivitást a sejtkultúrában szemben a kezeletlen H9c2 populációval, de a 100 µM PJ34 előkezelés esetén csak egy tendenciózus növekedést tapasztaltunk. A PARP aktiváció erőteljesebb blokkolásával növekvő védelmet várnánk. A magasabb koncentráció kísérleteinkben azonban nem javítja a további jótékony hatást, illetve a PrestoBlue esetén ez csak egy nem szignifikáns növekedést jelent. Így, azt gondoljuk, hogy 10 µM-os tartományú PJ34 teljes mértékben gátolja a PARP enzimet a H9c2 sejtekben 1 óra elteltével. Az az eredmény, hogy a magasabb koncentrációjú PJ34 nem javítja szignifikánsan a posztiszkémiás sejtek metabolikus aktivitását azt tükrözheti, hogy a magasabb koncentrációjú PARP gátlók metabolikus szupressziót okozhatnak, azonban ennek a hipotézisnek igazolásához további vizsgálatok szükségesek.
Következtetéseinket korlátozza, hogy a két populáció együttes LDH és metabolikus aktivitás szintjét mértük. Azonban ezeket a mérési módszereket gyakran használják a sejtalapú terápiák vizsgálatában és azt gondoljuk, hogy a két csoportot jellemző értékek értékes adatokat adnak tanulmányunkhoz.
További, a vizsgálatot limitáló tényezők:
- Vizsgálatainkat in vitro végeztük, ezen vizsgálati módszer minden előnyével és hátrányával. Hátrányt jelent, hogy a folyamatokat (pl. immunológiai) nem tudtuk teljes komplexitásában megjeleníteni/vizsgálni. Ugyanakkor előnye a módszerünknek, hogy standardizálható: a paraméterek (I-R ideje, hőmérséklet, O2 koncentráció) könnyen kontrollálhatók, a vizsgálat megismételhető, a terápiás és a posztiszkémiás sejtek mennyisége meghatározható. A 24 óra kokultivációs időt korábbi kísérleteinkre alapoztuk, ekkor már elegendő az idő a sejtek közötti kapcsolatok kialakulására, de még nem történik sejtproliferáció.
- H9c2 sejtvonallal dolgoztunk. A váz izom vagy patkány szívizom sejtek mellett ezt a sejtvonalat is gyakran használják hasonló kísérletekben [31, 38]. A terápiás sejtjeink szintén H9c2 sejtvonalból kerültek ki, amely nem őssejt és nem humán sejtvonal. Ugyanakkor korábbi kísérleteink kimutatták, hogy az egészséges H9c2 sejtek javítják a szomszédos, oxidatív stresszen átesett sejtek túlélőképességét. Sőt, a hasonló eredmény a H9c2 és az őssejtek adása esetén azt tükrözi, hogy nem szükségesek multipotens sejteket adnunk a terápiás hatás eléréséhez, ami kiszélesítheti a sejtterápiához szükséges sejtforrást.
Eredményeink összhangban állnak az eddigi, előkezeléssel kapcsolatos irodalmi adatokkal. Yao és mtsai [21] lipopoliszachariddal előkezelt mesenchymalis őssejteket adtak in vivo patkányokba. Jobb vaszkularizációt, kisebb mértékű fibrózist találtak a myocardiumban, és a terápiás sejtek túlélőképessége is nőtt. Hahn és mtsai [22] növekedési faktorral előkezelt sejteket használtak és a sejtek életképességét 30 perces 0,5%-os hypoxia után vizsgálták. 20%-os növekedést figyeltek meg. Ez a hatás mennyiségileg hasonló az általunk is talált PJ34 gátlószerrel elért eredményünkhöz a H2O2 modellünkben. Kubo és mtsai a csontvelő eredetű mesenchymalis őssejteket H2O2-vel 30 percen keresztül előkezelve a vaszkuláris endothelialis növekedési faktor szintjének növekedését és az endothelsejt felé történő differenciációt figyelték meg [20]. Mangi és mtsai a terápiás sejtek retrovirális úton kialakított Akt1 túlexpresszióját állította be, s ez a szív hatékonyabb működéséhez vezetett [23]. Ugyanakkor ismereteink szerint a mi vizsgálatunk az első kvantitatív életképességi adatokat felmutató vizsgálat, amelyben a terápiás sejtek előkezelése a posztiszkémiás sejteken lévő változást is bemutatja.
Összefoglalva tehát a PARP-inhibitorral előkezelt sejtek hozzáadása súlyosan károsodott szívizomsejtekhez javíthatja a túlélést és csökkentheti a nekrózist egy in vitro sejtterápiás iszkémia-reperfúzió modellben. Ez a megközelítés – ha humán sejtes, illetve in vivo vizsgálatokban is megerősítést nyer - hatékonyabb sejtalapú terápiához vezethet egy viszonylag egyszerű és gazdaságos előkezelési folyamattal.
Köszönetnyilvánítás
Jelen tanulmány az OTKA 83803, TÉT-SIN, TÁMOP 4.2.2-08/1/KMR-2008-0004, TÁMOP-4.2.1 / B 09/1/KMR- 2010-0001 támogatásával készült el.
Irodalom
- European cardiovascular disease statistics 2008. [cited 2011; Available from: http://www.heartstats.org/datapage.asp?id=7683.
- Cselenyak, A., et al., Stem cell transplantation in an in vitro simulated ischemia/reperfusion model. J Vis Exp, 2011(57): p. e3575.
- Dill, T., et al., Intracoronary administration of bone marrow-derived progenitor cells improves left ventricular function in patients at risk for adverse remodeling after acute ST-segment elevation myocardial infarction: results of the Reinfusion of Enriched Progenitor cells And Infarct Remodeling in Acute Myocardial Infarction study (REPAIR-AMI) cardiac magnetic resonance imaging substudy. Am Heart J, 2009. 157(3): p. 541-7.
- Medicetty, S., et al., Percutaneous Adventitial Delivery of Allogeneic Bone Marrow Derived Stem Cells Via Infarct Related Artery Improves Long-term Ventricular Function in Acute Myocardial Infarction. Cell Transplant, 2011.
- Paul, D., S.M. Samuel, and N. Maulik, Mesenchymal stem cell: present challenges and prospective cellular cardiomyoplasty approaches for myocardial regeneration. Antioxid Redox Signal, 2009. 11(8): p. 1841-55.
- Penn, M.S., et al., Stem cells for myocardial rege - neration. Clin Pharmacol Ther, 2011. 90(4): p. 499-501.
- Abdel-Latif, A., et al., Adult bone marrow-derived cells for cardiac repair: a systematic review and metaanalysis. Archives of internal medicine, 2007. 167(10): p. 989-97.
- Dayan, V., et al., Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic Res Cardiol, 2011.
- Ramos, G.A. and J.M. Hare, Cardiac cell-based therapy: cell types and mechanisms of actions. Cell Transplant, 2007. 16(9): p. 951-61.
- Gnecchi, M., et al., Paracrine mechanisms in adult stem cell signaling and therapy. Circulation research, 2008. 103(11): p. 1204-19.
- Plotnikov, E.Y., et al., Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in coculture. J Cell Mol Med, 2008. 12(5A): p. 1622-31.
- Cselenyák, A., et al., Mesenchymal stem cells rescue cardiomyoblasts from cell death in an in vitro ischemia model via direct cell-to-cell connections. BMC Cell Bio, 2010. 11: p. 29.
- Wu, K.H., et al., Stem Cell Engraftment and Survival in the Ischemic Heart. Ann Thorac Surg, 2011.
- Singla, D.K., et al., TGF-beta2 treatment enhances cytoprotective factors released from embryonic stem cells and inhibits apoptosis in infarcted myocardium. Am J Physiol Heart Circ Physiol, 2011. 300(4): p. H1442-50.
- Singla, D.K. and D.E. McDonald, Factors released from embryonic stem cells inhibit apoptosis of H9c2 cells. Am J Physiol Heart Circ Physiol, 2007. 293(3): p. H1590-5.
- Copland, I.B. and J. Galipeau, Death and inflammation following somatic cell transplantation. Semin Immunopathol, 2011. 33(6): p. 535-50.
- Lichtenauer, M., et al., Secretome of apoptotic peripheral blood cells (APOSEC) confers cytoprotection to cardiomyocytes and inhibits tissue remodelling after acute myocardial infarction: a preclinical study. Basic Res Cardiol. 106(6): p. 1283-97.
- CE Murry, e.a., Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature Reports Stem Cells, 2004(428): p. 664-8.
- M Koyanagi, e.a., (10): p. . Cell-to-Cell Connection of Endothelial Progenitor Cells With Cardiac Myocytes by Nanotubes: A Novel Mechanism for Cell Fate Changes? Circ Res, 2005(96): p. 1039-1041.
- Kubo M, L.T., Suzuki R, Ohshima M, Qin SL, Hamano K, Short-term pretreatment with low-dose hydrogen peroxide enhances the efficacy of bone marrow cells for therapeutic angiogenesis. Am J Physiol Heart Circ Physiol, 2007. 292: p. H2582-H2588.
- Yao, Y., et al., Lipopolysaccharide preconditioning enhances the efficacy of mesenchymal stem cells transplantation in a rat model of acute myocardial infarction. J Biomed Sci, 2009. 16: p. 74.
- Hahn, J.Y., et al., Pre-treatment of mesenchymal stem cells with a combination of growth factors enhances gap junction formation, cytoprotective effect on cardio - myocytes, and therapeutic efficacy for myocardial infarction. J Am Coll Cardiol, 2008. 51(9): p. 933-43.
- Mangi AA, N.N., Kong D, et al., Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. . Nat Med, 2003. 9: p. 1195-1201.
- László Virág, C.S., The Therapeutic Potential of Poly(ADP-Ribose)Polymerase Inhibitors. Pharmacol Rev, 2002(54).
- Pieper AA, W.T., Wei G, Clements EE, Verma A, Snyder SH, and Zweier JL Myocardial postischemic injury is reduced by polyADPripose polymerase-1 gene disruption. Mol Med, 2000(6): p. 271-282.
- J Zhang, V.D., TM Dawson, SH Snyder, Nitric oxide activation of poly(ADP-ribose) synthetase in neuro - toxicity. Science 1994(263): p. 687-689.
- Virag, L. and C. Szabo, The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev, 2002. 54(3): p. 375-429.
- M Haddad, H.R., C Bloquel, B Coqueran, C Szabó, M Plotkine, D Scherman, I Margaill, Anti-inflammatory effects of PJ34, a poly(ADPribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. British Journal of Pharmacology 2006. 149: p. 23-30.
- A Iwashita, N.T., S Matsuura, S Yamazaki, K Kamijo, J Ishida, H Yamamoto, K Hattori, N Matsuoka, S Mutoh, A Novel and Potent Poly(ADP-Ribose) Polymerase-1 Inhibitor, FR247304 (5-Chloro-2-[3-(4-phenyl-3,6- dihydro-1(2H)-pyridinyl)propyl]-4(3H)-quinazolinone), Attenuates Neuronal Damage in in Vitro and in Vivo Models of Cerebral Ischemia. JPET, 2004. 310: p. 425-436.
- C. Fiorillo, V.P., L Giannini, C Cecchi, A Celli, N Nassi, L Lanzilao, R Caporale, P Nassi, Protective effects of the PARP-1 inhibitor PJ34 in hypoxic-reoxygenated cardiomyoblasts. Cell Mol Life Sci, 2006(63): p. 3061-3071.
- Sardao, V.A., et al., Vital imaging of H9c2 myoblasts exposed to tert-butylhydroperoxide--characterization of morphological features of cell death. BMC cell biology, 2007. 8: p. 11.
- D Del Rio, A.S., N Pellegrini, A rewiew of recent studies on malondialdhyde as toxic molecule and biological marker of oxidative stress. Nutrition, Metabolism and Cardiovascular Diseases, 2005. 15: p. 316-328.
- Palma, P.F., et al., Evaluation of annexin V and Calcein-AM as markers of mononuclear cell apoptosis during human immunodeficiency virus infection. The Brazilian journal of infectious diseases : an official publication of the Brazilian Society of Infectious Diseases, 2008. 12(2): p. 108-14.
- Pankotai, E., et al., The role of mitochondria in direct cell-to-cell connection dependent rescue of postischemic cardiomyoblasts. Mitochondrion, 2011.
- Uemura, R., et al., Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Circulation research, 2006. 98(11): p. 1414-21.
- Oliveira, N.G., et al., Effect of poly(ADP-ribosyl)ation inhibitors on the genotoxic effects of the boron neutron capture reaction. Mutat Res, 2005. 583(1): p. 36-48.
- Vinod, K.R., S. Chandra, and S.K. Sharma, Evaluation of 5-aminoisoquinoline (5-AIQ), a novel PARP-1 inhibitor for genotoxicity potential in vitro and in vivo. Toxicol Mech Methods, 2010. 20(2): p. 90-5.
- Kimes, B.W. and B.L. Brandt, Properties of a clonal muscle cell line from rat heart. Experimental cell research, 1976. 98(2): p. 367-81.
Kapcsolat
Dr. Kiss Levente
Klinikai Kísérleti Kutató- és Humán Élettani Intézet,
Semmelweis Egyetem,
Tűzoltó utca 37-47.
Budapest, H-1094
Telefon: +36 20 3845753, FAX: +36 1 334 3162,
E-mail: Ez az e-mail-cím a szpemrobotok elleni védelem alatt áll. Megtekintéséhez engedélyeznie kell a JavaScript használatát.
Érbetegségek: 2001/3. 41-50. oldal