A közlemény új megvilágításba helyezi az érrendszerről alkotott korábbi ismereteinket. "Endothel szervről" beszél, melynek ép struktúrája és funkciója biztosítja az egész vasculatura integritását. Ebből a szempontból a thrombocyták adhéziója, aggregációja, az intrinsic véralvadás és a spontán librinolysis az integritást védő mechanizmusok.
Az atheroselerosis a vascularis dezintegritás legjellemzőbb formája. Az ér makroszkópos geometriai és mikroszkópos strukturális átalakulásával, azaz vascularis remodelinggel jár. A szerző leírja a pathomorfológiai változásokat és tárgyalja ezek klinikai konzekvenciáit.
Érbetegségek: 1994/2. 1-6. oldal
KULCSSZAVAK
vascularis integritás, vascularis remodeling, atheroselerosis
I. Endothel szerv
A vérkeringés élettani feladata a szervek tápanyag- és oxigénszükségletének biztosítása. Az aktív pumpa funkciót teljesítő szív mellett (sokáig úgy tűnt, hogy) az érrendszer szerepe csupán passzív transzport funkció, amely legfeljebb a nyomás-, áramlás-, perifériás rezisztencia változtatása révén befolyásolja a szervek aktuális igényeinek kielégítését. A vasculatura azonban nem passzív csőrendszer, hanem aktívan működő struktúra, amelynek belső rétege, az endothelium külön orgánumnak, önálló szervnek tekinthető. Az endothelium azért önálló szerv, mert teljesíti mindazokat a kritériumokat, amelyek a szervekre jellemzőek:
- Strukturálisan: azonos sejtek építik fel, a sejtek egymással érintkeznek, egymáshoz fekszenek, szövetet alkotnak. E szövet különlegessége, hogy csupán monocelluláris rétegből áll. Kiterített felülete hatalmas, mintegy 150- 1000 m , [1, 2], súlya pedig átlagosan 1,5 kg [3],
- Funkcionálisan: az endothelium szelektív barrier funkciója és autocrin- paracrin funkciója említendő. Ez utóbbi azt jelenti, hogy az endothel sejtek különböző anyagokat termelnek, amelyek lokálisan az endothel sejtek vagy közvetlen környezetük működését szabályozzák [2]. Az endothel sejtek által termelt anyagok [2, 4] három csoportba sorolhatók (1. ábra):
- A vasculatura tónusát befolyásoló anyagok:
Az EDRF (endothelial derived relaxing factor [1]), amely főként a NO- dal azonos, ez a legelektívebb vasodilatator. Az Endothelin-1, a vasoconstrictióért felelős molekula. - Antithromboticus anyagok:
PGI-2 (prostacyclin; [1, 5]), amely a fiziológiás thrombocyta aggregációt gátló anyag.
AT-III (antithrombin III; [1]), amely a fiziológiás alvadásgátló anyag.
t-PA (szöveti plasminogen aktivátor [1, 9]), amely a fiziológiás fibrinolyticus molekula. - Immunkompetenciáért felelős anyagok:
- IL-1 (interleukin-1) és egyéb ellenanyagok [1].
- A felsorolás nem teljes, hiszen a bradykinin, alfa2 makroglobulin, PAI (plasminogen activator inhibitor), von Willebrand faktor, növekedési faktorok (PDGF, platelet derived growt factor) vagy a MCP (monocyta chemotacticus protein [5]) stb. termelését is részben endothel sejtek végzik.
- Az endothelium tehát strukturális és funkcionális szempontból egyaránt önálló szervnek tekinthető.
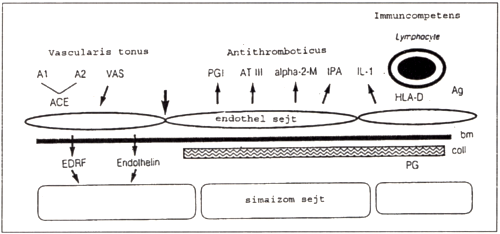
1. ábra
Endothel által termelt autocrin-paracrin anyagok.
II. A vasculatura integritását védő mechanizmusok
Az endothelium, vagyis az "endothel szerv" [3, 5, 6] védi az egész érrendszer strukturális és funkcionális épségét, vagyis védi a vasculatura integritását [3], A vasculatura integritása azon mechanizmusokat jelenti, amelyek az érrendszer strukturális és funkcionális épségét garantálják.
Az erek sérülésének három módja lehetséges:
- sérülhet az endothel;
- megnyílhat az érpálya és vérzés keletkezhet;
- a lumen elzáródhat.
Ennek megfelelően a vascularis integritásnak is három kiemelt jelentőségű mechanizmusa van:
- a thrombocyták adhéziója, aggregációja,
- az intrinsic véralvadás és
- a spontán fibrinolysis.
Thrombocyta funkciók
Sérülhet valahol az ér fala. Endothel sejtek sérülése többé-kevésbé elkerülhetetlen haemodynamikai jelenség, hiszen a keringés során a nyomás- és áramlásváltozások, a turbulencia, valamint a heterogén összetételű vér viscositásából adódó belső súrlódás sérthetik az endotheliumot, és lokális fali károsodást eredményezhetnek. Az endothelsejtek által termelt anyag, a von Willebrand faktor [5, 7, 8, 28] azonban aktiválja a thrombocytákat, azok adhéziója, kisebb aggregációja, úgynevezett muralis thrombus [5, 6, 9] jelentkezik. Az összecsapódott vérlemezkék testükkel fedik be az endothel sérülést, lehetővé téve a neointima kialakulását, az endothel reparatióját. A thrombocyták élettani funkciója tehát a sérült endothel reparatiója (sajnos sokszor túlméretezett reakcióval).
Intrinsic véralvadás
Nagyobb sérülés esetén megnyílhat az érpálya, vérzés keletkezhet. A vérzés gyors haemodynamikai katasztrófával fenyeget, miközben a nagyobb sérülés, vagyis az ér falán keletkezett nyílás befedéséhez a thrombocyták adhéziója és aggregációja már elégtelen.
Az intrinsic véralvadás akkor jelentkezik, ha a vér érintkezik a perivascularis szövetekkel. Normálisan intravascularis coagulatio nincs. Az érpályát elhagyó vér azonban - érintkezve a környező szövetekkel - megalvad. Úgynevezett "haemostaticus dugó" [8, 10] alakul ki, mely elzárja az intimát, médiát és adventitiát is perforáló nyílást, elősegítve a gyógyulást. Az intrinsic véralvadás tehát az érrendszer sérülésének alapvető reparatios mechanizmusa (sajnos sokszor túlméretezett reakcióval).
Spontán fibrinolysis
Az erek károsodásának harmadik formája a lumen elzáródása, melyet egyrészt a keringéssel odajutó, illetve az ér falán és falában lerakódott anyagok, másrészt thrombus, embolus, illetve ezek együttesen okozhatnak. A coagulum alapvető tényező az occlusio kialakulásában.
Az endothel sejtek által termelt t- PA (szöveti plasminogen aktivátor [11, 12] a keringésben lévó, plasminogen plasminná alakulását segíti elő. A plasmin fibrinolyticus molekula [11], amely oldja a fibrinszálat és oldja az alvadékot is. A klinikumban időnként megfigyelhető spontán recanalisatio a t-PA inducalta spontán fibrinolysisnek tulajdonítható. A spontán fibrinolysis is az érsérülés reparatiójának fontos tényezője.
Összefoglalva: a thrombocyták funkciója, illetve az intrinsic véralvadás és a spontán fibrinolysis a vasculaturát védő és integritását fenntartó alapvető mechanizmusok.
III. Atherosclerosis - vascularis remodeling
A korábban ép endothelium - elvesztve eddigi szelektív barrier funkcióját - strukturálisan és funkcionálisan dezintegrálódik, és sejtek (monocyták, T lymphocyták), molekulák (LDL), bizonyos anyagok (Ca) számára átjárhatóvá válik [5], Ez az a folyamat, amit érelmeszesedésnek, atherosclerosisncik hívunk, és ami a vasculatura dezintegritásának leggyakoribb és legpregnánsabb reprezentánsa. Az atherosclerosis az ér makroszkópos geometriai és mikroszkópos strukturális átalakulásával jár. Ezt az átalakulást vascularis remodelingnek nevezzük [13, 14], Az ér geometriai átalakulását a lumen szűkülete, az érfal megvastagodása, esetleg kitágulása jelzi, a strukturális átrendeződést pedig a monocyták és simaizomsejtek migratioja, proliferatiója, phenotypus (funkció) változása és necrosisa képezi. Ezen négy jelenség a a vascularis remodeling sejtbiológiai alapja [13, 14], Nem az atherosclerosis a vascularis remodeling egyetlen, de mégis a leggyakoribb formája. Az atherosclerosis sokáig klinikai konzekvenciák nélkül zajló jelenség. Az endothel sérülését követően [5], esetleg anélkül is [15, 16, 17] az endothel sejtek felszínére jutó specifikus adhéziós glycoproteinek [5] és egy endothel által termelt anyag, az MCP (monocyta chemotacticus protein) hatására a monocyták kitapadnak az endothelium felszínére [18, 19], (2. ábra) Megállnak a keringésben, mintegy odatapadnak az endothel sejtekhez. Az ilyen monocyták később az endothelsejtek rései között per diapedesim [20] átbújnak a subintimalis térbe.
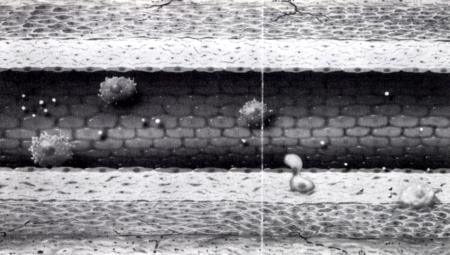
2. ábra
Atherosclerosis pathomechanizmusa (I): a monocyták megállnak a keringésben, odatapadnak az endothel sejtekhez, majd per diapedesim az endothel sejtek között átbújnak a subintimalis térbe és macrophággá alakulnak.
Az átjutást deformabilitásuk segíti, vagyis e sejtek - a higanycsepphez hasonlóan - képesek keskeny nyílásokon alakváltozással átbújni. A subintimalis térbe jutó monocyták phenotípusa megváltozik, macrophaggá alakulnak [21] és proliferálnak. A macrophagok feladata a subintimalis térbe került anyagok eltakarítása, azaz igazi takarító, scavenger funkció. A macrophagok scavenger sejtek. Elsősorban a lumenből a subintimalis térbe jutó zsírmolekulák, oxidált LDL molekulák eltakarítását végzik. Nem tudjuk, pontosan miért és milyen endocytosison keresztül kerül az LDL molekula (cholesterin) a subintimalis térbe, miért válik az endothel e zsírmolekulák számára könnyen átjárhatóvá, azaz miért veszti el barrier funkcióját, hogyan internalizálódik [20], esterificalódik, és hol történik az LDL molekula oxidálódása [5, 23, 25, 26, 27, 28, 29], legfeljebb annyi bizonyos, hogy e jelenség a "túlkínálat", azaz magas serum cholesterin esetén gyakrabban megfigyelhető. A subintimalis térben a macrophagok phagocytálják a zsírmolekulákat, és úgynevezett habos sejtekké (foam cell) válnak (3. ábra).
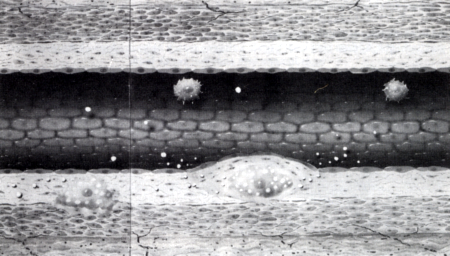
3. ábra
Atherosclerosis pathomechanizmusa (II): A macrophágok scavenger sejtek, a subintimalis térben phagocytálják az LDL molekulákat és habos sejtté (foam cell) válnak.
A habos sejtek accumulatiója sárgás, csíkos elszíneződést okoz az intimán, és ez az atherosclerosis első látható megjelenése, a "fatty streak" [20, 30, 31, 32, 33]. Természetesen később a habos sejtek elpusztulhatnak, széteshetnek és a phagocytált zsírmolekulák ismét szabadon a subintimalis térbe kerülhetnek [34], E jelenségek nem maradnak hatás nélkül a média simaizom sejtjeire sem: a myocyták elhagyják a rnediat, a subintimalis térbe vándorolnak [16], és proliferalnak [35]. Feladják eddigi contractilis funkciójukat és szintetikus funkciójúvá, azaz szintetikus phenotípusúvá válnak. Elsősorban collagen rostokat kezdenek szintetizálni. A myocyták tevékenysége tehát alapvetően megváltozik, a korábbi contractio helyett migratio, proliferatio és szintézis lesz alapvető funkciójuk. Az átalakulás ingerét a PDGF (platelet derived growth factor [5, 36, 37, 48])-nak nevezett növekedési factor képezi, melyeket a thrombocytákon kívül endothel sejtek és a macrophagok is termelnek.
Végülis olyan elváltozások jönnek létre, anielyeket a széli részeken elhelyezkedő thrombocyták és macrophagok, centrálisán elhelyezkedő habos sejtek és extracelluláris zsír jellemez, körben pedig myocyták, illetve az általuk termelt fibrosus kötőszövet, colla- gen (részben elastin) vesz körül. Ezt az elváltozást atheroscleroticus (fibrosus) plaque-nak nevezzük, mert az intimalis felszínből kiemelkedik és a lumen több-kevesebb stenosisát eredményezi (4. ábra). Később Calcium depositio révén a plaque elmeszesedhet.
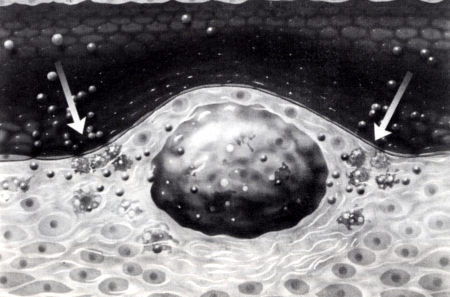
4. ábra
Atherosclerosis pathomechanizmusa (III): A simaizom sejtek is a subintimalis térbe vándorolnak és kollagént (elastint) szintetizálnak. A kialakult atheroscleroticus plaque közepén szabad zsír és habos sejtek, széli részein macrophágok, körben simaizom sejtek, kollagén rostok helyezkednek el. Az elváltozást az ér lumene felé endothel sejtek fedik.
A történet mindezideig prae-, vagy subklinikai, azaz klinikai tünetekkel nem jár, és megjelenése évekkel, évtizedekkel megelőzheti a releváns klinikai manifesztációkat. Klinikai tünetek ugyanis akkor keletkeznek, ha az atheroscleroticus plaque növekedése miatt már áramlási akadályt is okoz. Klinikai tünetekhez 75%-nál nagyobb szűkület szokott vezetni (stabil angina pectoris, cerebrovascularis tünetek, claudicatio intermittens). A plaque rendszerint merev, és környezeténél kevésbé képes rugalmasan alkalmazkodni az érfal feszülésváltozásaihoz. Ezért vérnyomásingadozás vagy áramlási turbulencia a plaque széli részein berepedéshez [38, 39, 40], rupturához, majd intraplaque haemorrhagiához vezethet (5. ábra).
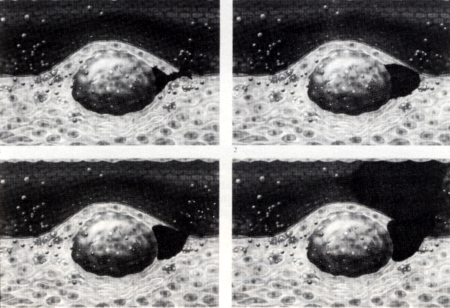
5. ábra
Atherosclerosis pathomechanizmusa (IV): Acut ischaemiás syndroma: plaque ruptura, intraplaque haemorrhagia, melyet thrombus képződés és a lumen hirtelen elzáródása követ.
Természetesen mindez a plaque hirtelen növekedését eredményezi [41, 42], amely harsány klinikai tünetek (instabil angina pectoris vagy infarktus, stroke, alsóvégtagi acut ischaemiás syndroma) jelentkezésével jár. Az acut vascularis események hátterében tehát rendszerint az atheroscleroticus plaque rupturája és bevérzése áll. Miután sérült az endothel, a vasculatura integritását védő mechanizmusok működésbe lépnek. A thrombocyták adhéziója, aggregációja és az intraplaque haemorrhagiát követő intrinsic véralvadás igyekszik a sérülést helyreállítani. Túlméretezett reakciójuk miatt azonban coagulum nemcsak a plaque-ban jelentkezik, hanem a lumenre is ráterjed, és a megduzzadt atheroscleroticus plaque mellett a coagulum a lument végleg elzárja [11, 13]. A t-PA feloldhatja az alvadékot a plaque-ban és a lumenben egyaránt, így megnyithatja az elzárt lument. Az endothel sérülés helyét neo intirna [5, 44, 45, 46, 47,48] tüntetheti el, sőt az atheroscleroticus plaque regressziójára is vannak bizonyítékok. Calcium lerakódás révén meszes köpeny fedheti, takarhatja be az elváltozást. Vagyis a rupturált plaque részben gyógyulhat is, sokszor azonban a történet kimenetele más irányú, inkább pathológiai és klinikai romlást eredményez.
Atheroscleroticus plaque és plaque ruptura, folyamatos progresszió és partialis regresszió, stabil és instabil klinikum váltakozása, reverzibilis tünetek és irreverzibilis sérülések jellemzik az atherosclerosis pathológiáját és klinikumát, jellemzik a vasculatura dezintegritását, azaz az atherosclerosissal járó vascularis remodelinget.
Irodalom
- Camilleri, J. P. Hypertension and arterial aging (Les Laboratories Servier) Springer Verlag. 1994. 1-84.
- Camilleri, J. P., Berry, C. L., Fies- singet, J. N. et al: Diseases of the arterial wall. Springer Verlag (1989)
- Towards vascular integrity. Sparre Medical, Sweden. Sandoz Pharma Ltd., Basel kiadványa. (1994)
- The atherosclerotic process in the hypertensive patient. Symposium highlights. 1992. VI. 12. Toledo, Spain. Pfizer kiadvány (1992)
- Ross, R.: Hypothesis of atherogenesis. In: Schlant R. C" Alexander R. W. The Heart, arteries and veins. 8 Ed. McGraw-Hill Inc. Health Pro- fessions Divison New York, St. Louis. San Francisco 994-1008. (1994)
- French, J. E.: Atherosclerosis in relation to the structure and function of the arterial intima, with special reference to the endothelium. Int. Rev. Exp. Pathol. 5: 253-353. (1966)
- Jaffe, E. A., Hoyer, L. W., Nachman R. I.; Synthesis of antihemophilic factor antigén by cultured humán endothelial cells: J. Clin. Invest 52: 2757-2764. (1973)
- Sixma, J. .J., Wester, J.: The hemostatic plug. Sémin. Hematol. 14: 265-299. (1977)
- Kessler, C. M.: Anticoagulation and Thrombolytic therapy. Chest. 95: Suppl. 245S-256S. (1989)
- Marder, V. J., Sherry, S.: Thrombolytic therapy. Current status. NEJM. 318: 1512-1520. (1988)
- Loscalzo, J.: An overview of thrombolytic agents. Chest. 97: Suppl. 117-123. (1990)
- Van Hinsbergli, V. W. M, Sprengers, E. D., Kooistra, T.: Effect of thrombin on the production of plasminogen activators and PI-1 by humán foreskin microvascular endothelial cells. Thromb. Hae- mostas. 57: 148-153. (1987)
- Gibbons, G. H" Dzau, V. J.: Mechanisms of disease: The emerging concept of vascular remodeling. NEJM. 330: 1431-1438. (1994)
- Ross, R.: The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362: 801-809. (1993)
- Ross, R.: Atherosclerosis: a problem of the biology of arterial wall cells and their interactions with blood components. Arteriosclero- sis 1: 293-311. (1981)
- Ross, R.: The pathogenesis of arteriosclerosis - an update. NEJM. 314: 488-499. (1986)
- Valente, A. .J., Graves, D. T., Vialle-Valentin, C. E. et al: Purification of a monocyte chemotactic factor (SMC-CF) secreted by non human primate vascular smooth muscle cells in culture. Bioche- mistry. 27: 4162-4168. (1988)
- Joris, J., Zand, T., Nannari, J. et al: Studies on the pathogenesis of atherosclerosis adhaesion and emigration of mononuclear cells in the aorta of hypercholesterolaemic rats. Amer. J. Pathol. 113: 341- 358. (1983)
- Gerrity, R. G.: The role of the monocyte in atherogenesis I. Transiti- on of bloodborne monocytes into foam cells in fatty laesions. Amer. J. Pathol. 103: 181-190. (1981)
- Ross, R.: The pathogenesis of atherosclerosis. In Braunwald E. ed Heart Disease: a textbook of car- diovascular medicine 4th ed vol. 2. Philadelphia. W. B.: Saunders 1106-1124. (1992)
- Schwartz, C. J., Valente, A. J., Sprague, E. A. et al: The pathogenesis of atherosclerosis: an overview. Clin. Cardiol. 14: 1-16. (1991)
- Stein, Y, Stein, 0. Interaction between serum lipoproteins and cellular components of the arterial wall. Biochem, Atherosclerosis: 7: 313- 344. (1979)
- Rosenfeld, M. E., Lipton, B. A.: Cellular responses to oxidized LDL. Curr. Opin. Lipidol. 3: 318- 323. (1992)
- Carew, T. E., Schwenke, D. C., Steinberg, D.: Antiatherogenic effect of probucol anrelated to its hypocholesterolemic effect: evidence that antioxidants in vivo can selectively inhibit low density lipoprotein degradation of atherosclerosis in the Watanabe hereditable hyperlipidemic rabbit: Proc. Natl. Acad. Sci. USA. 84: 7725-7729. (1987)
- Simionescu, N., Simionescu, M., Palade, G. E.: Permeability of muscle capillaries to small hemopeptides: Evidence for the existence of patent transendothelial chan- nels. J. Cell. Biol. 64: 586-590. (1975)
- Renkin, E. M.: Multiple pathways of capillary permeability. Circ. Res. 41: 735-743. (1977)
- Witzum, J. L., Steinberg, D.: Role of oxidised LDL in atherogenesis. J. Clin. Invest. 88: 1785-1792. (1991)
- Rimm, E. B., Stempfer, M. .J., Ascherio, A. et al: Vitamin E con- sumption and the risk of coronary heart disease in men. NEJM. 328: 1450-1456. (1993)
- Stampfer, M. J., Hennekens, C. H., Manson, J. E. et al: Vitamin E consumption and the risk of coronary heart disease in women. NEJM. 328: 1444-1449. (1993)
- WHO Classification of atherosclerotic lesions. Report of a study group. WHO Technical Report Se- ries. 143: 1-20. (1958)
- Guzman, M. A., McMahan, C. A., McGill, H. C. et al: Selected methodologic aspects of the International Atherosclerosis Project. Lab. Invest. 18: 479-497. (1968)
- Faggiotto, A., Ross, R., Harker, L.: Studies of hypercholesterolaemia in the non-human primate I. Changes that lead to fatty streak formation. Arteriosclerosis. 4: 323-340. (1984)
- Faggiotto, A., Ross, R.: Studies of hypercholesterolaemia in the nonhuman primate II. Fatty Streak conversion to fibrous plaque. Arteriosclerosis. 4: 341-356. (1984)
- Geer, J. C., McGill, H. C., Strong, J. P.: The fine structure of human atherosclerotic lesions. Amer. J. Pathol. 38: 263-277. (1961)
- Davies, M.: Pathogenesis of atherosclerosis (Review): Curr. Opon. Cardiol. 7: 541-545. (1992)
- Ross R" Glomset J. A., Kariya B. et al.: Platelet dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc. Natl. Acad. Sci. USA. 71: 1207-1210. (1974)
- Gajdusek, C., Di Corleto, P., Ross R. et al: An endothelial cell deri- ved growth factor. J. Cell. Biol. 85: 467-472. (1980)
- Davies, M. .J., Thornas, A. C.: Pathologic basis and microanatomy of occlusive thrombus formation in human coronary arteries. In Interactions between Platelets and Vessel Walls. Edited by. Born G. V. R" Vane J. R. London. Royal Society. 9-12. (1981)
- Falk, E.: Plaque rupture with severe preexisting stenosis precipitating coronary thrombosis: characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br. Heart. J. 50: 127- 134. (1983)
- Richardson, P. D., Davies, M. J., Born, G. V. R.: Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet 11: 941-944. (1989)
- Levin, D. C., Follon, J. T.: Significance of the angiographic morphology of localised coronary stenoses. Histopathological correlates: Circulation. 66: 316-320. (1982)
- Falk, E.: Unstable angina with total outcome: dynamic coronary thrombosis leading to infarction and/or sudden death. Circulation. 71: 699-708. (1985)
- Barger, A. C., Beenwkes, III., R., Lainey, L. L. et al: Hypothesis: vasa vasorum and neovascularisation of humán coronary arteries. NEJM. 310: 175-177. (1984)
- Wissler, R. W., Vesselinovitch, D. Studies of regression of advanced atherosclerosis in experimentál animals and man. Ann. N. Y. Acad. Sci. 275: 363-378. (1976)
- Blankenhorn, D. H., Nessitn, S. A., Johnson, R. L. et al.: Beneficial effects of combined colestipol-niacin therapy on coronary atherosclerosis and coronary venous bypass grafts. JAMA. 257: 3233-3240. (1987)
- Brown, B. G., Albers, J. J" Fischer, L. D. et al: Regression of coronary artery disease as a result of intensive lipid lowering therapy in men with high levels of apolipo- protein B. NEJM. 323: 1289-1298. (1990)
- Wissler, R. W., Vesselinovitch, D.: Can atherosclerotic plaques reg- ress? Anatomical and biochemical evidence from nonhuman animal models: Amer. J. Cardiol. 65: (Suppl) 33-40. (1990)
- Blankenhorn, D. H.: Can atherosclerotic lesions regress? Angiographic evidence in humans. Amer. J. Cardiol. 65: Suppl. 41-43. (1990)
Dr. Káli András
Országos Kardiológiai Intézet
1096 Budapest, Haller u. 29
A cikk illusztrációinak közlését a SANDOZ cég budapesti képviselete tette lehetővé.
Érbetegségek: 1994/2. 1-6. oldal